

Introduction
Avocado (Persea americana Mill.) is today among the most economically important subtropical/tropical fruit crops in the world (Bost, Smith, & Crane, 2013), with a production of avocado fruit that now exceeds 3.5 million tons, of which about 20 % is traded internationally (Schaffer, Wolstenholme, & Whiley, 2013). Chanderbali et al. (2008) consider avocado as the most important commodity from the Lauraceae.
The conservation of avocado genetic resources and their relatives is important to deal with the potential problems of the avocado industry in the future. Threats to the avocado industry have appeared recently, such as laurel wilt, caused by the fungus Raffaelea lauricola symbiont of the ambrosia beetle (Xyleborus glabratus) that has been responsible for the extensive death of native Lauraceae in the United States since 2000, when it was first detected (Fraedrich et al., 2008). In August 2011, a dooryard avocado tree immediately north of the focus was affected by laurel wilt (Ploetz et al., 2015), close to the center of avocado production in Florida, USA. Resistance to this disease is now of high priority; the pool to search for this resistance is in the genetic resources of the genus Persea.
Germplasm banks have tried to conserve the existing diversity of avocado and its relatives (Barrientos, 2010), one of them located in the Fundación Salvador Sánchez Colín-CICTAMEX, S.C., which is considered the richest in respect to diversity and variability, and which started to concentrate more diversity in 1988 (Barrientos, 1999). The variability of this germplasm bank has been reported (López-López, Barrientos-Priego, & Ben-Ya’acov, 1999), as well its potential (Ben-Ya’acov & Barrientos, 2003), along with molecular characterization of some accessions with RAPD (Reyes-Alemán, Valadez-Moctezuma, Simuta-Velázco, Barrientos-Priego, & Gallegos-Vázquez, 2013), ISSR (Reyes-Alemán, Valadez-Moctezuma, & Barrientos-Priego, 2016), SSR (Gutiérrez-Díez, Barrientos-Priego, & Campos-Rojas, 2015) and with the sequence trnL-trnF of cpDNA (Cabrera-Hernández et al., 2017). In these studies, the great variability existing in that germplasm bank was evident, where the accessions represent above all the diversity that exists in the subgenus Persea.
The knowledge of the phylogenetic relationships of the subgenus Persea with the subgenus Eriodaphne is important to take decisions in relation to management and organization of germplasm banks and to guide future collections, in addition to defining actions with respect to genetic improvement.
The genus Persea L. (Lauraceae) consists of about 85 species distributed in America (Barrientos-Priego, Muñoz-Pérez, Borys, & Martínez-Damián, 2015), some new species have been described (Lorea-Hernández, 2002; van der Werff, 2002) and there are probably over a 100 species. The genus is distributed from the southern United States (Persea borbonia [L.] Spreng) to Chile (Persea lingue Ruiz & Pavon), with one species in the Canary Islands (P. indica [L.] Spreng.) and probably some representatives in South Asia (Barrientos-Priego et al., 2015); nevertheless, it is controversial as to whether Persea should be treated as including species from Asia since results suggest that Persea is strictly American (Li et al., 2011). The genus is divided into the subgenera Persea and Eriodaphne (Kopp, 1966); the first one has fruits known as real avocados (~ 5 to 20 cm) and the second tiny avocados known as “aguacatillos” (< 5 cm).
Within subgenus Persea, P. americana Mill. is the most studied species, mainly for its importance as a human food resource, and especially for its high oil content. For these reasons, and considering the graft compatibility among species, attempts to use species of subgenus Eriodaphne as a rootstock for P. americana to improve resistance to Phytophthora cinnamomi Rands. have been explored; however, the unsuccessful results revealed a vegetative incompatibility between species of both subgenera (Frolich, Schroeder, & Zentmyer, 1958).
There is a great controversy about the monophyletic origin of the genus Persea, indicating that phylogenetic studies based on morphological characters are not conclusive (Rohwer et al., 2009), and the subgenera Persea and Eriodaphne might perhaps be recognized as independent genera. However, a recent study by Li et al. (2011) shows Persea as monophyletic again, if Apollonias is included and a few aberrant species excluded. Several studies of the Lauraceae family based on molecular data give some information about Persea phylogeny (Chanderbali, van der Werff, & Renner, 2001); nevertheless, the inclusion of few species and specimens made the results uninformative for the Persea-Eriodaphne clade. The subgenus Eriodaphne has been studied by sequencing fragments of nuclear and chloroplast DNA more extensively by other authors (Chanderbali et al., 2001; Li et al., 2011; Rohwer et al., 2009), while the subgenus Persea has not. Cabrera-Hernández et al. (2017) in their study indicated that other sequences (chloroplast, mitochondrial and nuclei) must be studied in a concatenated way to have a better resolution of the subgenus Persea.
Specifically, within Persea, the cladistic analysis of Campos-Rojas, Terraza, and López-Mata (2007), the ITS phylogenetic study of Rohwer et al. (2009) and the trnL-trnF of cpDNA study of Cabrera-Hernández et al. (2017) could separate into different clades the species of the subgenus Persea from the species of Eriodaphne, supporting the hypothesis of a polyphyletic origin of the genus Persea, and providing an explanation of the vegetative (Frolich et al., 1958) and gametic (Lahav & Lavi, 2013) incompatibility between the two subgenera. However, controversy still exists on this issue, because the phylogenetic relationships between the two subgenera are very complex (Kopp, 1966), and so far, there is insufficient evidence from molecular DNA data for the separation of the two subgenera of Persea.
In several families of angiosperms, DNA sequences of coding regions, intergenic spacers and internal transcribed spacers of the chloroplast, mitochondria, and nucleus have been used in a concatenated form to obtain a better understanding of the phylogenetic relationships of the taxa analyzed. Among the most used genes are: rbcL (Kress & Erickson, 2007), ndhF (Beilstein Nagalingum, Clements, Manchester, & Mathews, 2010), matK, rpoC1 (Chase et al., 2007), and the intergenic spacer region trnH-psbA (Dong, Liu, Yu, Wang, & Zhou, 2012) from chloroplast DNA. Also, fragments of mitochondrial DNA, such as atp4 gene (Duminil, Pemonge, & Petit, 2002), and the nuclear 18S rRNA gene have been considered. With these novel analyses, it is evident that information from different DNA genes of several Persea species is necessary to reconstruct the phylogenetic history of this genus. For this reason, the study aims to analyze the phylogenetic relationships within the genus Persea, with an emphasis on the subgenus Persea, using maximum parsimony and bayesian inference with the sequence of eight different fragments from nuclear, chloroplast and mitochondrial DNA.
Material and methods
Plant material
Plant material from 35 specimens of the genus Persea, 29 of Persea subgenus and five of Eriodaphne subgenus, and one from Beilschmiedia anay (Blake) Kosterm, were obtained from Fundación Salvador Sánchez Colín-CICTAMEX, S.C. germplasm bank (Coatepec Harinas, Mexico), and from specimens deposited at the herbarium of the Forestry Department at Universidad Autónoma Chapingo, Mexico (CHAP). The specimens are from locations inhabited by the genus in Mexico and other countries (Table 1). The accessions included in the study represent practically all the diversity (seven species) of the subgenus Persea, according to the Kopp (1966) classification, although the unrecognized species Persea zentmayerii is not included (Schieber & Bergh, 1987). In the case of Persea americana, all races or botanical varieties were included, as well as the proposed fourth race Persea americana var. costaricensis. In addition, some hybrids were considered (Table 1), as well as Beilschmiedia anay that was used as an outgroup.
Table 1.
|
|
||||||||||
| CG-Hu-56 | Puebla, Mexico | |||||||||
| CHAP 37473Z | Hidalgo, Mexico | |||||||||
| CH-C-30 | Michoacan, Mexico | |||||||||
| CH-Pl-1 | Chile | |||||||||
| CH-G-36 | Veracruz, Mexico | |||||||||
|
|
CH-PR-1 | Veracruz, Mexico | ||||||||
|
|
||||||||||
|
|
CH -CR- 28 | Costa Rica | ||||||||
|
|
CH-G-48 | Yucatán, Mexico | ||||||||
|
|
CH-G-45 | Yucatán, Mexico | ||||||||
|
|
CH-I-6 | Veracruz, Mexico | ||||||||
|
|
‘Hass’ | California, U.S.A. | ||||||||
|
|
CH-CR-25 | Costa Rica | ||||||||
|
|
CH-CR-44 | Costa Rica | ||||||||
|
|
CH-C-10 | Puebla, Mexico | ||||||||
|
|
CH-C-47 | Michoacan, Mexico | ||||||||
|
|
CH-C-57 | Mexico, Mexico | ||||||||
|
|
CH-C-63 | Mexico, Mexico | ||||||||
|
|
CH-Der-2 | Mexico, Mexico | ||||||||
|
|
CH-G-7 S2 | Chiapas, Mexico | ||||||||
|
|
CH-G-11 S1 | Chiapas, Mexico | ||||||||
|
|
CH-GU-5 | Guatemala | ||||||||
|
|
CH-GU-6 | Guatemala | ||||||||
| CH-I-3 | Veracruz, Mexico | |||||||||
|
|
CH-I-2 | Mexico, Mexico | ||||||||
| CH-G-76 | Chiapas, Mexico | |||||||||
| CH-I-4 | Israel | |||||||||
| CH-Ve-2 | Veracruz, Mexico | |||||||||
| CH-Der-1 | Veracruz, Mexico | |||||||||
| CH-Gu-1 | Guatemala | |||||||||
| CH-H-5 | Honduras | |||||||||
| CH-H-7 | Honduras | |||||||||
|
|
CH-C-62 | Guatemala | ||||||||
| CH-G-Ch1 | Chiapas, Mexico | |||||||||
| Mv1 | Chiapas, Mexico | |||||||||
|
|
CH-CR-29 | Costa Rica | ||||||||
DNA extraction, amplification, and sequencing
DNA was extracted from ~ 50 to 100 mg of leaves previously dried in silica gel. In some cases, leaves from herbarium specimens were used. Genomic DNA was extracted by the cetyltrimethylammonium bromide (CTAB) based method (Gambino, Perrone, & Gribaudo, 2008). At the end of the procedure, the DNA was purified with Qiaquick columns (Qiagen®, USA) following manufacturer's instructions. The quality and quantity of the DNA were evaluated with a NanoDrop® ND-1000 spectrophotometer. The amplification of each of the eight fragments was performed in a total volume of 25 µL containing: 50 to 100 ng of DNA, 200 µM of dNTPs mix, 1X Colorless GoTaq® Flexi Reaction Buffer (Promega, USA), 20 pM of specific primers (Table 2), 2.5 mM of MgCl2 and 2 U of GoTaq® Flexi DNA Polymerase (Promega, USA). Amplification programs consisted of one cycle of an initial denaturation of 4 min at 94 °C, followed by 35 cycles of 45 s at 94 °C, 1 min at specific melting temperature (Table 2) and 1 min at 72 °C, finally an extension of 5 min at 72 °C. The amplification reactions were performed in a GeneAmp® PCR System 9700 thermocycler (Applied Biosystems, USA).
Table 2.
| Locus/segment | Name | Sequence 5’-3’ | Tm (°C) | Reference |
|---|---|---|---|---|
|
nz |
GTAGTCATATGCTTGTCTC | 56 |
White, Bruns, Lee, & Taylor ( |
|
| CTTCCGTCAATTCCTTTAAG | 56 | White et al. (1990) | ||
| AACTTAAAGGAATTGACGGAAG | 56 | White et al. (1990) | ||
| TCCGCAGGTTCACCTACGGA | 56 | White et al. (1990) | ||
| cp |
1f | GTGGATACACTTCTTGATAATGG | 56 | Ford et al. (2009) |
| 4r | TGAGAAAACATAAGTAAACGGGC | 56 | Ford et al. (2009) | |
| cp |
trnH2 | CGCGCATGGTGGATTCACAATCC | 51 | Tate & Simpson (2003) |
| psbAF | GTTATGCATGAACGTAATGCTC | 51 | Tate & Simpson (2003) | |
| cp |
1f | ATGTCACCACAAACAGAAAC | 56 | Olmstead, Michaels, Scott, & Palmer (1992) |
| 724r | TCGCATGTACCTGCAGTAGC | 56 | Fay, Swensen, & Chase (1997) | |
| cp |
389f | CTGCBACCATAGTMGCAGCA | 59 | This study |
| 461r | GATTRGGACTTCTRSTTGTTCCGA | 59 | This study | |
| cp |
1326R | TCTAGCACACGAAAGTCGAAGT | 48 | Schmitz-Linneweber et al. (2001) |
| 390F | CGATCTATTCATTCAATATTTC | 48 | Schmitz-Linneweber et al. (2001) | |
| mt |
Orf1 | AAGACCRCCAAGCYYTCTCG | 50 | Duminil et al. (2002) |
| Orf2 | TTGCTGCTATTCTATCTATT | 50 | Duminil et al. (2002) | |
| mt |
Cox3r | CTCCCCACCAATAGATAGAG | 51 | Duminil et al. (2002) |
| Cox3f | CCGTAGGAGGTGTGATGT | 51 | Duminil et al. (2002) |
The amplified DNA fragments were visualized on a 1.2 % agarose gel stained with ethidium bromide. The polymerase chain reaction (PCR) products were cleaned using Qiaquick® PCR Purification Kit columns (Qiagen, USA), following the instructions provided by the manufacturer. The PCR products were sequenced directly using the same primers (Table 2) in an automated sequencing system in Macrogen Inc., South Korea. The sequences were edited and assembled with the BioEdit version 7.0.9.0 program (http://www.mbio.ncsu.edu/BioEdit/bioedit.html).
Sequence alignment
The 34 sequences obtained from the intergenic spacer trnH-psbA, ndhF, rbcL, rpoC1, 18S rRNA, cox3, and atp4 genes, and 35 from the matK gene (Table 2) were aligned with MUSCLE version 3.8 (Edgar, 2004). Additionally, 16 sequences of matK were aligned with 36 sequences downloaded from GeneBank (http://ncbi.nlm.nih.gov): two of Persea and 18 from the closely related genera (Sassafras, Litsea, Lindera, Ocotea, Cinnamomum, Nectandra, Actinodaphne, Parasassafras, Sinosassafras, Neolitsea, Iteadaphne, Endlicheria, Aniba, Laurus, Umbellularia, Alseodaphne, Phoebe and Machilus). Afterward, two super-matrices, the first one with the chloroplast DNA sequences: ndhF + rbcL + matK + rpoC1 + trnH-psbA and the second with all eight, were built manually.
Phylogenetic analysis
The 52 aligned sequences of matK, and the two super-matrixes mentioned above were analyzed with maximum parsimony (MP) using PAUP ver. 4.0b10 software (Swofford, 2001) and bayesian inference (BI) using MrBayes ver. 3.1.2 (Ronquist & Huelsenbeck, 2003). The mitochondrial genes and the nuclear rDNA data were not analyzed separately since they did not show sufficient informative characters. In each analysis of MP, all the characters were weighted equally, and gaps treated as missing data. A set of the most parsimonious trees from the different datasets was obtained through heuristic searches of 1,000 replicates with random stepwise sequence addition, tree bisection-reconnection branch (TBR) swapping, ‘‘MulTrees’’ option in “effect”, and saving 10 trees from each random sequence addition. Robustness of clades was estimated by a bootstrap analysis with 1,000 replicates with simple sequence addition, TBR swapping and holding only 10 trees per replicate to reduce time spent in swapping on large numbers of suboptimal trees. The BI was performed using the GTR + G model and two independent replicates of four chains with a maximum of 10 million generations, with trees sampled every 100 generations.
Results
Features of the sequence alignments
A total of 273 sequences were obtained from ndhF, rbcL, matK, rpoC1, trnH-psbA, 18S rRNA, atp4 and cox3; all of them were deposited at GenBank under Accession numbers JF966395-JF966399, JF966401-JF966414, JF966416-JF966418, JF966420, JF966423-JF966426, JF966428-JF966672, and JQ352803 (Table 1). The trnH-psbA alignment held the highest variation, with 32 parsimony-informative sites (Pi, 6.44 %), and 67 variable sites (VS, 13.48 %) (Table 3). The mitochondrial genes atp4 and cox3 held the least variation, with 0 to 1 Pi sites, and 0.18 and 0.43 % VS, respectively (Table 3); despite the low informative sites obtained, it was decided to include them. Beilschmiedia anay CG-Hu-56 had the most divergent sequence in the eight sequences, by a variation of 0-4 % with P. americana sequences. B. anay CG-Hu-56 was used as an outgroup in the phylogenetic analysis.
Table 3.
| Locus/segment | Alignment length (bp) |
CR |
NCR | Pi (%) | CS (%) | VS (%) | S | EFM |
|---|---|---|---|---|---|---|---|---|
| n |
1748 | 0 | 1748 | 6 (0.34) | 1719 (98.34) | 29 (1.69) | 23 | 2 |
| cp |
599 | 599 | 0 | 2 (0.33) | 577 (96.33) | 22 (3.67) | 20 | 2 |
| cp |
497 | 98 | 399 | 32 (6.44) | 428 (86.12) | 67 (13.48) | 41 | 5 |
| cp |
1481 | 1428 | 53 | 10 (0.67) | 1390 (93.86) | 91 (6.14) | 81 | 4 |
| cp |
739 | 739 | 0 | 4 (0.54) | 707 (95.67) | 32 (4.33) | 28 | 0 |
| cp |
909 | 909 | 0 | 7 (0.77) | 866 (95.27) | 43 (4.73) | 36 | 1 |
| mt |
507 | 507 | 0 | 1 (0.20) | 501 (99.82) | 6 (1.18) | 5 | 0 |
| mt |
695 | 695 | 0 | 0 (0.00) | 692 (99.57) | 3 (0.43) | 3 | 0 |
|
|
4236 | 3773 | 463 | 55 (1.30) | 3965 (93.60) | 261 (6.16) | 206 | 12 |
| 18S rRNA+ |
7183 | 4983 | 2200 | 62 (0.86) | 6874 (95.69) | 299 (4.16) | 237 | 14 |
Phylogenetic analysis of matK
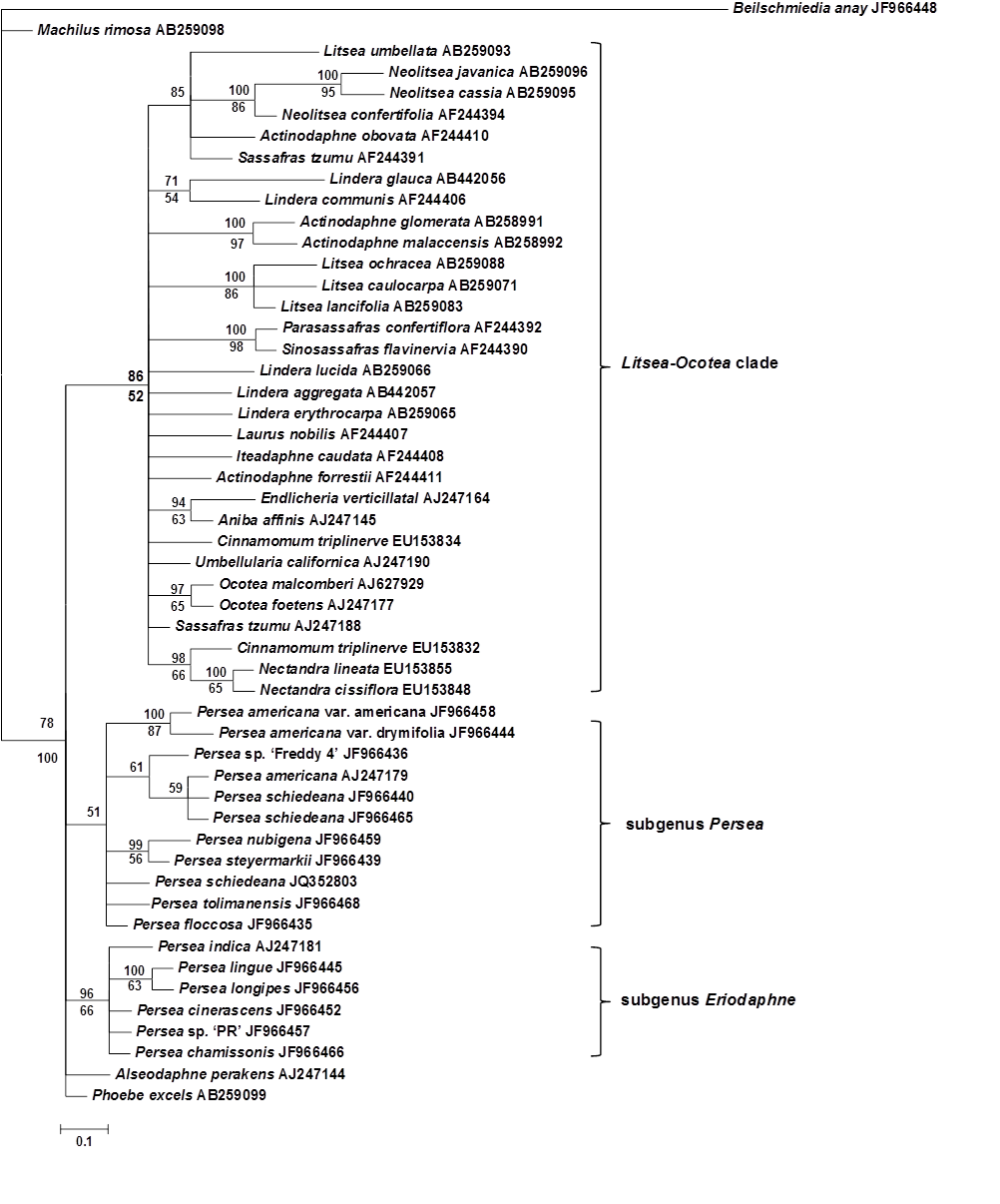
A large phylogenetic analysis was performed with the matK. To place the subgenera Persea and Eriodaphne inside the Lauraceae family, representatives of 18 closely related genera were included in the analysis. Both the BI and the MP approaches resulted in relatively congruent topologies concerning subgenus Eriodaphne and the Litsea-Ocotea clade, and although Persea subgenera species were grouped with a weak Posterior Probability (PP) in BI, the bootstrap (BS) majority rule consensus tree from MP does not support this clade (Figure 1). The MP and BI recovered the subgenus Eriodaphne and the Litsea-Ocotea clade with weak BS and strong PP, BS values for these clades are 52 and 66 %, and BI support for the same branches is 86 and 96 %, respectively. Within the Eriodaphne clade, both analyses support the subclade P. lingue-P. longipes, with 63 and 100 % of BS and PP, respectively. In the Litsea-Ocotea clade, both analyses support the formation of eight different subclades, mainly with species of the same genera, with 63 to 98 % of BS values and 71 to100 % of PP (Figure 1). Beilschmiedia anay JF966448 and Machilus rimosa AB259098 are separated from the main core (100 % PP).
Analysis of the concatenated chloroplast sequences
The phylogenetic analysis of the five chloroplast sequences was performed with sequences of 34 different plant accessions evaluated in this study, with members of the subgenera Persea and Eriodaphne, plus Beilschmedia anay. The BI and MP analyses resulted in relatively congruent topologies (Figure 2). The analyses recovered two major clades, subgenus Eriodaphne and subgenus Persea, with well-supported BS/PP (88/100 %) and moderate values (82/84 %), respectively. This indicates that the additional parsimony informative characters from the other chloroplast sequences may have improved the phylogenetic signal.

On the other hand, the five genes have a total of 261 VS, with 22 in rpoC1 to 91 of rbcL; of these, 55 are Pi sites, with two in rpoC to 32 in trnH-psbA (Table 3). Also, it is important to note the presence of 12 fixed mutations in the five species of subgenus Eriodaphne so far investigated, which have led to the formation of a very solid clade (Table 3).
Within the five accessions of the subgenus Eriodaphne clade, the BI supports two groups, in the MP-BS majority rule consensus tree, although just the Persea chamissonis-Persea sp. ‘PR’ clade has a weak support of 61 %. This clade was also supported in the matK analysis. Within the Persea clade, there was a basal polytomy of two accessions of species of Persea americana (var. americana, CH-G-45 from Yucatán, Mexico and var. guatemalensis CH-G-11 S1 from Chiapas, Mexico), Persea parvifolia (CH-Ve-2 from Veracruz, Mexico) and a clade comprising the rest of the accessions (Figure 2). In this subclade, the BI tree shows five clades; two of them strongly supported one with all the Persea americana var. drymifolia accessions and another with Persea nubigena CH-I-4, Persea steyermarkii CH-G-Ch1 and P. tolimanensis Mv1; one with weak support; another with negligible; plus, one consisting of the single Persea floccosa CH-I-3 (Figure 2).
Analysis of the eight concatenated sequences
The phylogenetic analysis of the eight sequences was performed with plant accessions of 29 members of the subgenus Persea, five of the subgenus Eriodaphne and Beilschmiedia anay. The BI and MP analyses also resulted in relatively congruent topologies (Figure 3), and in general very similar to the BI and MP tree of the concatenated chloroplast sequences. The subgenera Eriodaphne and Persea clades were also obtained, but with slightly higher BS/PP support, 94/100 % for Eriodaphne and 84/86 % for Persea (Figure 3). The addition of 18S rRNA, cox3, and atp4 genes provided 38 VS, seven of which are Pi (Table 3). This information was not able to significantly improve the phylogenetic signal. The Eriodaphne fixed mutations increased from 12 to 14, by the addition of two mutations of the 18S rRNA gene (Table 3).

Discussion
Persea is one of the most complex genera of the Lauraceae. Previous phylogenetic analyses of the matK gene (Chanderbali et al., 2001; Rohwer, 2000; Rohwer et al., 2009) have shown that the Persea group is a monophyletic group deeply nested within the Lauraceae, close to the Litsea and Ocotea complexes. In previous analyses, such as the trnL-trnF/trnH-psbA phylogenetic tree of Chanderbali et al. (2001), both subgenera of Persea are grouped in the same clade, related to Machilus thunbergii and Alseodaphne semecarpifolia. In the ITS phylogeny of Chanderbali et al. (2001), the three species of subgenus Eriodaphne formed a small clade (97 % BS), with Persea americana as its immediate sister group and several other, mainly Asian species of the Persea group as sister group to both. However, the small number of specimens analyzed of the two subgenera did not allow resolving the relationships within the Persea group.
Rohwer et al. (2009) used ITS sequences of several genera of the family. They found that the species of the subgenera Persea and Eriodaphne grouped separately from each other and from Machilus species. In our study, although matK gene showed a low degree of divergence in the sequences analyzed, BI and MP phylogenies could set the subgenus Eriodaphne in an independent clade, separated from species of the subgenus Persea and the other genera analyzed. Rohwer (2000) also found low levels of divergence within sequences of matK in Lauraceae (9.7 %) and less than 1 % within the genus Persea.
Although the trnH-psbA spacer region and the rbcL gene are more variable than matK (Table 3), these genes were not selected to investigate the position of Persea within the Lauraceae, because the trnH-psbA intergenic spacer has two areas subjected to frequent inversions that are not analyzed in this study and the phylogenetic trees of the rbcL (not shown) had the same topologies as the trees of matK.
The trees obtained from the analysis of chloroplast sequences and the eight concatenated ones are almost the same, due to the 55 PI sites of the chloroplast sequences, making them the most useful for the phylogenetic reconstruction of the clades, especially for the subgenus Persea. The mitochondrial and 18S rRNA genes only contributed to the separation of two accessions of Persea schiedeana (CH-H-5 and CH-Gu-1), although with moderate support.
In the subgenus Eriodaphne all species considered were resolved completely, but in the subgenus Persea the analysis failed to separate Persea americana from all the species, especially from Persea schiedeana, which has also been found in a study of avocado germplasm and additional species of subgenus Persea with ISSR markers (Reyes-Alemán et al., 2016). The genetic variability level of the avocado, despite its cross-pollination system, is not considered to be exceptionally high compared with estimates that have been made with temperate fruit species (Chen, Morrel, de la Cruz, & Clegg, 2008), which seems to be what was found in part in the present study.
Persea parvifolia L. O. Williams (Persea pallescens [Mez] Loera-Hernández), a shrub with thin shoots, small narrow obovate to elliptic leaves and small fruits (Figure 4), which was first described by L.O Williams (1977) and not considered by van der Werff (2002) as a subgenus Persea species, is one of the most ancestral species in the subgenus Persea clade, so it could be considered as a good candidate for the species that gave rise to the avocado; however, it was unresolved with the other two individuals of P. americana that also have a conserved sequence, so they could be primitive forms of those races. More individuals of this species are needed for a further analysis as well as other P. americana and other sources of P. parvifolia to support this.

It has been indicated that although P. nubigena, P. steyermarkii and P. floccosa could be separated from P. americana by restriction fragment length polymorphism (RFLP), they are considered to be only variants of P. americana (Furnier, Cummings, & Clegg, 1990); however, the results show that some of these species cluster together, which is the case of P. nubigena, P. steyemarkii, and P. tolimanensis, species considered to contribute to the ancestry of P. americana var. guatemalensis (Schieber & Bergh, 1987); nevertheless, this does not seem to correspond to our findings.
With respect to P. americana, a well-supported clade that includes five accessions of the Mexican race (P. americana var. drymifolia) were grouped together with two of the West Indian one (P. americana var. americana) indicates that they are closely related. It can be assumed that the last two accessions are not completely pure and that they may have genetic characteristics of the Mexican race. Conversely, an apparent conflict between phenotypic and genotypic data can help adjust pedigree information (Ashworth & Clegg, 2003), and be used to reclassify accessions in the germplasm bank as possible hybrids. This last point also applies to another clade that grouped accessions of the Guatemalan race, possibly hybrid, one P. americana var. costaricensis, and a P. schiedeana from Honduras, the last of which was also reported using DFP and SSR markers which did not find unique DNA patterns which could characterize the three races of P. americana and the three accessions of P. schiedeana (Mhameed et al., 1997). This is also in accordance for the subclade that grouped two P. schiedeana, one from Honduras and the other from Guatemala. In the other subclade, two accessions of Costa Rica were together an unclassified one (‘Freddy 4’) and a P. americana var. americana (CH-CR-28), which is probably the West Indian Race subclade.
The complex legacy of ancient and recent avocado improvement has left a profusion of genotypes of uncertain affinities and with diffuse racial boundaries (Ashworth & Clegg, 2003), where other factors may have a role, including the possibility of remote hybridization events (Bufler & Ben-Ya’acov, 1992) or a more recent date for racial differentiation than previously thought (Ashworth & Clegg, 2003).
It must be considered that although the analyses of the eight concatenated sequences separate both subgenera of Persea, the variation of the eight sequences is low, 4.16 % of VS and 0.86 % of Pi sites (Table 3). This was reported for trnH-psbA (Chanderbali et al., 2001) and matK (Rohwer, 2000) in the family Lauraceae, but not for the other sequences. Therefore, it is necessary to find sequences showing a greater variation that allow a better resolution of the phylogenetic relationships within subgenus Persea. A suitable candidate may be the nuclear ITS region, which has 33 % parsimony-informative sites for many Lauraceae accessions (Rohwer et al., 2009), but in our experience it has the disadvantage of being difficult to amplify and sequence in some accessions of Persea, and to align because of too many indels. Liu, Chen, Song, Zhang, and Chen (2012) found that the ITS2 region produced a low success rate in direct PCR amplification and sequencing in Lauraceae species and it is also unsuitable to be the DNA barcode of the family.
Based on the hypothesis of a monophyletic origin of the genus Persea, our results partially suggest that this genus is not a monophyletic group; therefore, one could think that the subgenera Persea and Eriodaphne should be recognized as independent genera, confirming the analysis of Rohwer et al. (2009), where Persea does not appear to be monophyletic, because the subgenus Persea seems to be more closely related to Phoebe and Alseodaphne than to the subgenus Eriodaphne.
Conclusions
The eight concatenated sequences separated both subgenera (Persea and Eriodaphne) into two different clades, where 14 fixed mutations were found in the studied species of the subgenus Eriodaphne, supporting the hypothesis of independent genera. In the subgenus Persea, the concatenated sequences used failed to separate Persea americana from all the species, especially from Persea schiedeana, the most distinct species in the subgenus. The chloroplast intergenic spacer trnH-psbA sequence held the highest variation and informative sites, while the mitochondrial and nuclear rDNA sequences studied were not informative.